Para las distrofias musculares no miotónicas como la Distrofia de Duchenne y la Distrofia de Becker, la cardiopatía forma parte inherente de su espectro clínico. La expresión clínica de éstas se puede manifestar con insuficiencia cardíaca descompensada, arritmias o muerte súbita; gran parte de ellos cursan asintomáticos en el transcurso del tiempo, pero cuando se manifiestan constituyen una de las principales causas de muerte en estos pacientes, por lo que su detección temprana y tratamiento óptimo influyen en gran medida en el pronóstico clínico de estos pacientes. A continuación, se presenta el caso de un paciente en quien se encontró de forma incidental el compromiso cardíaco de uno de estos desórdenes neuromusculares.
Palabras clave: distrofia muscular, insuficiencia cardíaca, compromiso cardíaco, distrofina.
CARDIAC INVOLVEMENT IN MUSCULAR DYSTROPHIES: A CLINICAL CASE
For non-myotonic muscular dystrophies such as Duchenne Dystrophy and Becker Dystrophy, heart disease is an inherent part of its clinical spectrum. The clinical expression of these diseases can be manifested with decompensated heart failure, arrhythmias or sudden death; A large part of them are asymptomatic over time, but when they manifest they constitute one of the main causes of death in these patients, so their early detection and optimal treatment greatly influence the clinical prognosis of these patients. The following is the case of a patient in whom the cardiac involvement of one of these neuromuscular disorders was found incidentally.
Keywords: muscular dystrophy, heart failure, cardiac involvement, dystrophin.
CASO CLÍNICO
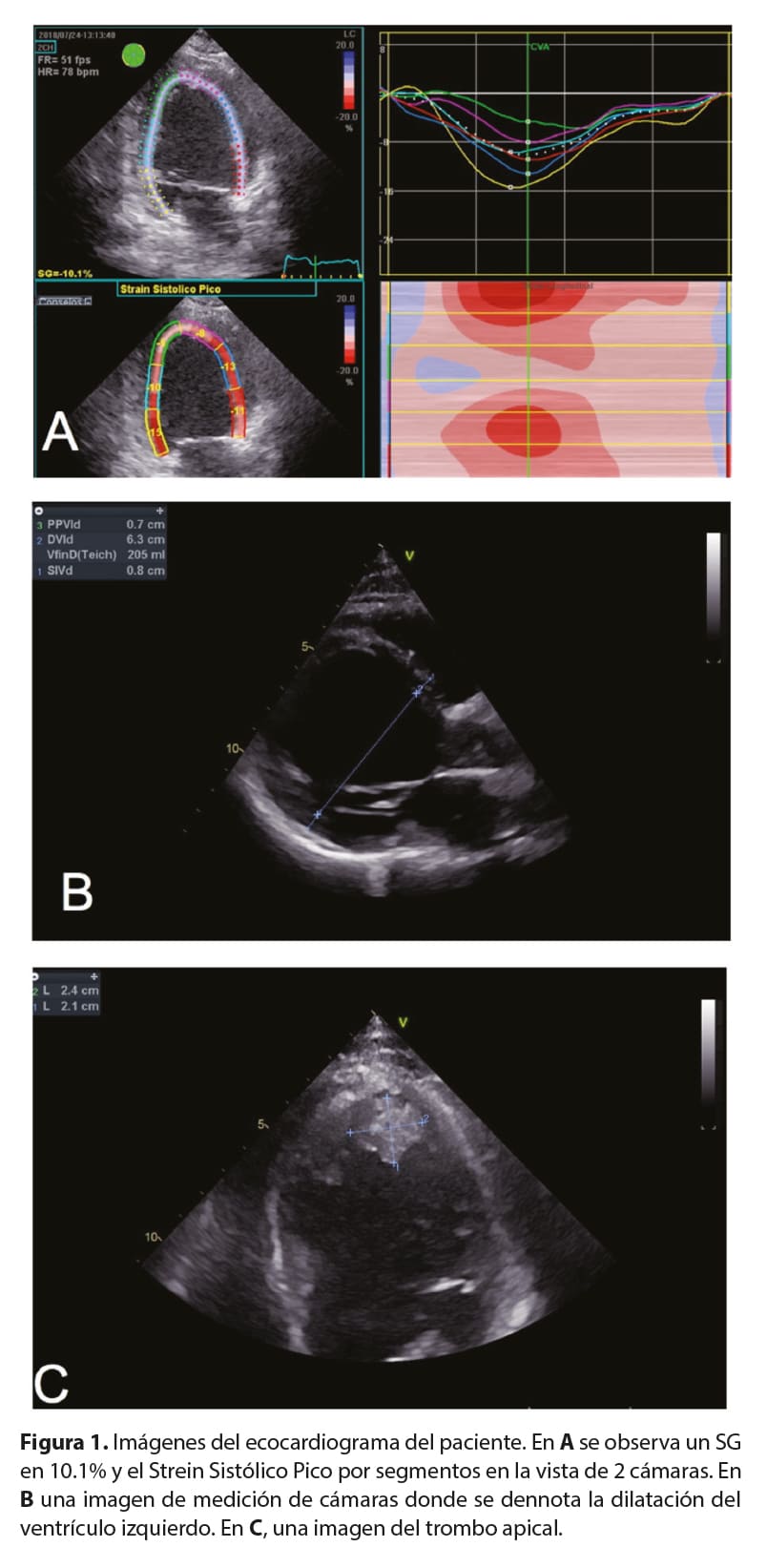
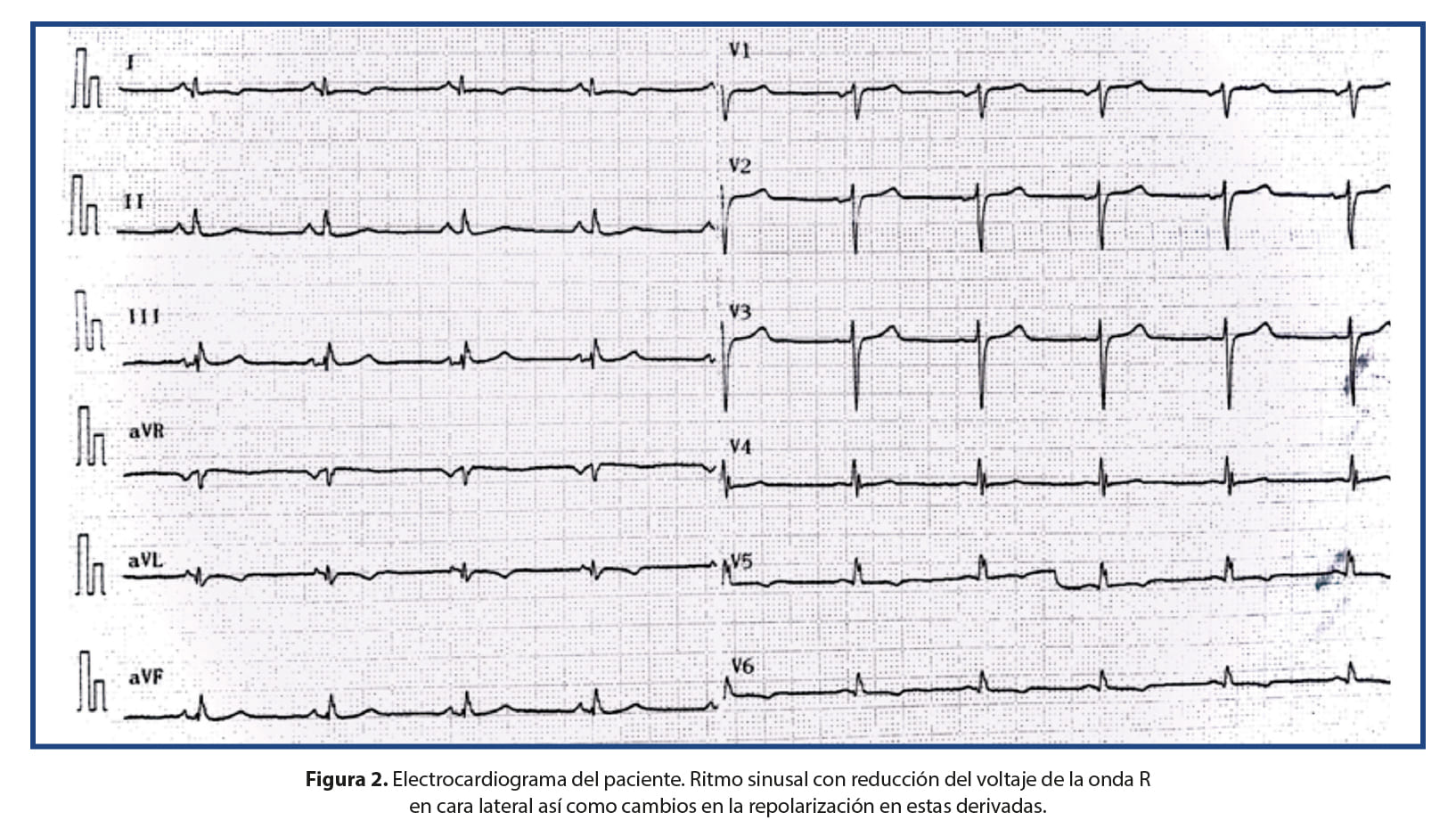
Se trata de un paciente masculino de 21 años, con historia familiar de distrofia muscular tipo Becker en un hermano menor, un primo y tres tíos maternos. Desde la infancia, ha padecido de mialgias, quien ha cursado asintomático desde el punto de vista cardiovascular. Quien ingresa con historia de un día de evolución de cefalea universal intensa, asociando luego debilidad en hemicuerpo izquierdo, documentándose un evento cerebrovascular tipo isquémico lacunar derecho. La Tomografía Axial Computarizada (TC) de cerebro al ingreso presentó un ASPECTS score de 10. Dentro de los estudios complementarios realizados al ingreso, se realiza un ecocardiograma que documenta una miocardiopatía en fase dilatada (Figura 1) con fracción de eyección del ventrículo izquierdo severamente deprimida, en 15%, y se documenta trombo apical. Al examen físico, llama la atención la hipertrofia de pantorrillas, a la auscultación cardíaca con taquicardia sinusal, sin soplos ni ruidos añadidos, rítmico; y sin datos de insuficiencia cardíaca. En el electrocardiograma presenta ritmo sinusal, con reducción del voltaje de la onda R en cara lateral así como cambios en la repolarización en estas derivadas (Figura 2). Dado estos hallazgos y la historia familiar del paciente, se decide realizar una biopsia endomiocárdica (Figura 3).
HALLAZGOS HISTOPATOLÓGICOS
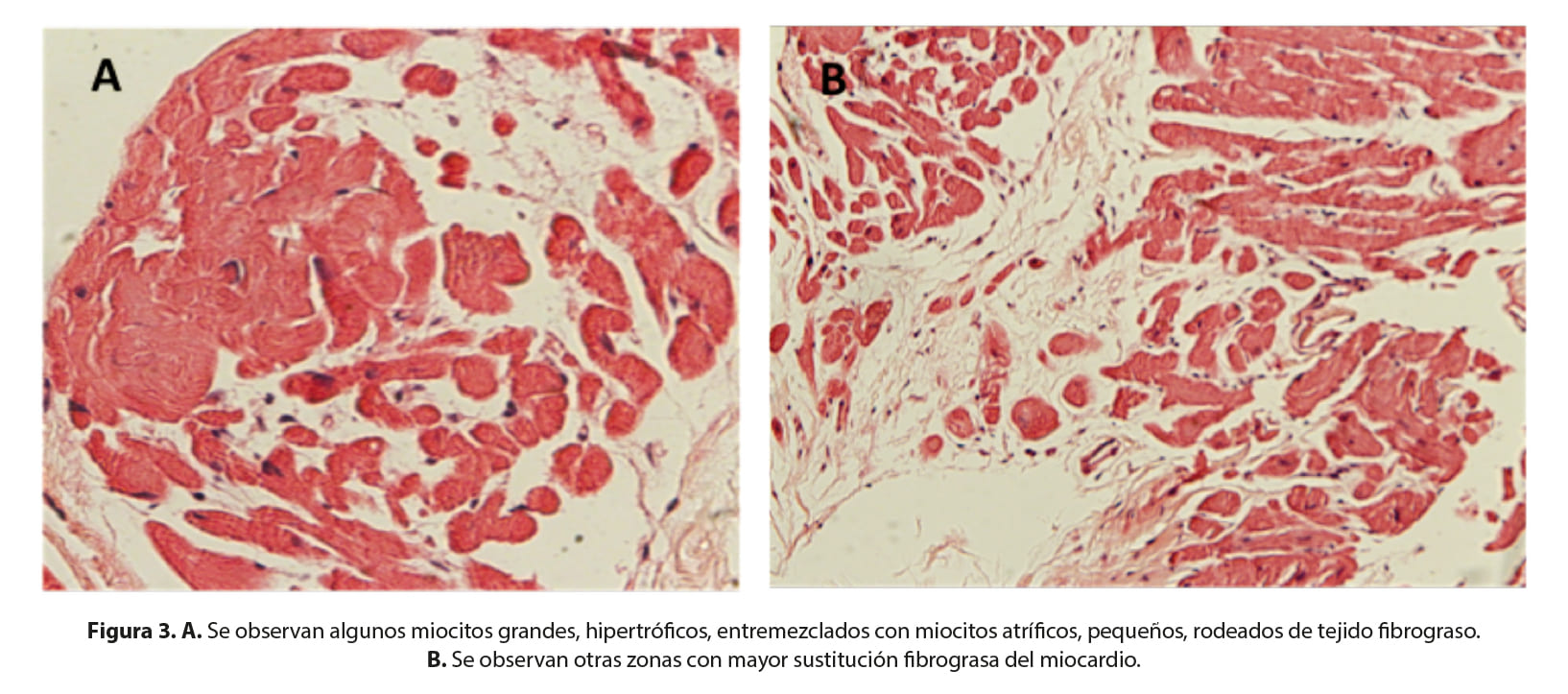
La muestra histológica está constituida por miocardio, en donde se observan fibras musculares grandes, de forma redondeada, algunas con centralización de los núcleos, compatibles con fibras hipertróficas (Figura 2). Éstas están asociadas a fibras pequeñas, anguladas, de aspecto atrófico, con abundante tejido fibrograso circundante, en algunas zonas sustituyendo al miocardio (Figura 2b). Estos hallazgos constituyen cambios miopáticos de tipo distrófico.
Las distrofias musculares comprenden un grupo de desórdenes neuromusculares que se presentan con debilidad muscular particularmente progresiva, de algunos grupos musculares que varían según la patología. La distrofia muscular de Becker y la de Duchenne son los dos tipos más representativos del grupo de las distrofias ligadas al cromosoma X1,2.
Ambas se caracterizan por mutaciones en el gen de la distrofina, una proteína importante para la estructura del complejo proteoglicano-distrofina que tiene funciones en la estabilización y señalización entre la membrana celular del miocito, la matriz extracelular y el citoesqueleto. En el caso de la distrofia de Duchenne, las mutaciones principales son deleciones o duplicaciones, que lleva a un cambio en el marco de lectura y a su vez a la ausencia total de la expresión de la distrofina o a una proteína disfuncional.
La distrofia muscular de Becker por su parte, se caracteriza por deleción de exones, es decir, una mutación genética en la que se pierde material genético de los exones que son los que llevan la codificación de la proteína distrofina; y duplicaciones de material genético, así como mutaciones puntuales o mutaciones que afectan el splicing, que conlleva a la eliminación de aminoácidos de la proteína final, lo cual causa que la distrofina en la histología se muestre reducida en cantidad o tenga un tamaño anormal pero aún funcional3.
Manifestaciones clínicas de las distrofias musculares
Las manifestaciones clínicas varían desde afectación de músculo esquelético, músculo cardíaco e incluso un 7.5% de los pacientes con el tipo Becker presentan epilepsia por afección cerebral y también retardo mental4. En el caso de la distrofia de Duchenne, los síntomas iniciales se presentan en la infancia temprana, con dificultad para correr y luego para subir escaleras, instaurada de forma progresiva. Muchos de estos pacientes no pueden caminar hacia los 12 años y la expectativa de vida ronda las 3 décadas1 .
Por su parte, la distrofia muscular de Becker es menos frecuente que la distrofia de Duchenne. En general, tiene manifestaciones clínicas más heterogéneas y una evolución más leve. En la primera década de vida, cursan sin manifestaciones clínicas relevantes pero sí con elevación de la creatin kinasa (CK) que se acompañan luego de anormalidades de la marcha que se caracterizan por un movimiento lateral de las caderas. Después de los 20 años, inicia una debilidad progresiva y la pseudohipertrofia de las pantorrillas puede encontrarse en algunos casos. En la tercera década inicia la dificultad para subir escaleras y realizar ciertos trabajos manuales, mialgias e incluso mioglobinuria episódica. Pueden tener una expectativa de vida de 5 o 6 décadas3,4.
Compromiso cardíaco de las distrofias musculares
El compromiso cardíaco es una característica frecuente en algunas distrofias, como en el caso de la distrofia muscular de Becker y constituye una de las principales causas de muerte en estos pacientes (la primera en el caso del tipo de Becker). Ocurre un reemplazo de la arquitectura normal de los miocitos cardíacos por cardiomiocitos disfuncionales, así como sustitución del tejido de conducción por tejido fibroso o grasa4. Hay un grupo de pacientes que puede ser asintomático o subclínico durante toda la vida. En un tercio de los casos, se trata de una miocardiopatía dilatada que desencadena luego falla cardíaca, siendo la edad promedio de afectación cardíaca los 28 años. En los casos sintomáticos, el espectro puede variar desde arritmias hasta muerte súbita, incluyendo también falla cardíaca descompensada4. Los pacientes pueden referir palpitaciones, mareos, síncope, disnea, o bien manifestarse abiertamente como falla cardíaca aguda descompensada. La severidad de la clínica a nivel de músculo esquelético no tiene correlación con el compromiso cardíaco o el tiempo de su aparición.
El mecanismo fisiopatológico del compromiso cardíaco en estos pacientes no se conoce aún en detalle, sin embargo en estudios con animales se ha sugerido que tiene relación con alteraciones tempranas en el metabolismo celular y transducción de señales1,3. Asimismo, un exceso de calcio intracelular y generación de especies reactivas de oxígeno, con la consecuente disrupción del potencial de membrana mitocondrial puede relacionarse con el daño inicial del sarcolema por deficiencia de distrofina y en la disfunción mitocondrial.
A nivel de laboratorio, se puede documentar aumento de CK y CK-MB, pero no son útiles para llegar al diagnóstico3. El electrocardiograma es de suma importancia en estos pacientes y se ha descrito que incluso en la mayoría de los casos, es anormal. El patrón clásico distintivo son las onda R altas y aumento de la amplitud R/S en V1 con ondas Q profundas y estrechas en las derivadas precordiales izquierdas4. También, no es infrecuente documentar acortamiento del PR sin onda delta, taquicardia sinusal, datos de hipertrofia ventricular derecha, fibrilación atrial y retraso de la conducción intraventricular con ensanchamiento del QRS. En algunos reportes de casos, se ha encontrado que la cara inferolateral es la principal afectada en la distrofia de Becker, en los que se ha documentado ondas Q significativas en derivaciones II, III, aVF y V6 que correlaciona con afección de la cara posterobasal y lateral; además de reducción del voltaje de la onda R.5,6 Se atribuye esta afección miocárdica a estrés mecánico aumentado y no tanto a la distribución de distrofina de esta zona.
Asimismo, el bloqueo incompleto de rama derecha del Haz de His es un hallazgo frecuente que se asocia con compromiso temprano del ventrículo derecho. Por su parte, el bloqueo de rama izquierda del Haz de His se puede encontrar en pacientes con cardiomiopatía dilatada4.
En cuanto a estudios de imágenes, se puede realizar ecocardiograma, PET, resonancia magnética e incluso biopsia endomiocárdica para valorar el compromiso histologico1. En el ecocardiograma, puede presentar engrosamiento miocárdico dependiendo del estadío de la enfermedad, que varía desde cavidades de tamaño normal con función sistólica preservada y en otra proporción, se encuentra una reducción de la fracción de eyección y dilatación del ventrículo izquierdo. La miocardiopatía hipertrófica usualmente va a evolucionar a miocardiopatía dilatada, con hipoquinesia global y función sistólica reducida, o bien, conservada3. Las alteraciones de la contractilidad segmentarias se ven principalmente en la pared posterior y lateral. Puede encontrarse también trombos apicales o insuficiencias valvulares secundarias3.
La resonancia magnética cardíaca, por su parte, nos brinda información no solo anatómica y estructural, sino también funcional. En este caso, se puede encontrar áreas de fibrosis miocárdica con el gadolinio de forma temprana durante la evolución de la enfermedad5. Actualmente, el seguimiento con estudios de imagen se recomienda idealmente con resonancia magnética cardíaca al menos cada 2 años después del diagnóstico en caso de no encontrar hallazgos patológicos, para la detección temprana de cardiomiopatía e iniciar el tratamiento lo antes posible para enlentecer la progresión de la disfunción cardíaca3.
Por su parte, la biopsia endomiocárdica puede revelar algunos cambios que dependerán de la fase o estadío en que se encuentre el paciente. En las fases más tempranas, hay hipertrofia de los miocardiocitos y fibrosis endomiocárdica o intersticial. Conforme avanza la enfermedad, puede encontrarse más fibrosis endomiocárdica. Es importante dennotar que no se han descrito características histológicas que permitan diferenciar una distrofia muscular de otra. Para diferenciar adecuadamente entre distrofia de Duchenne y una distrofia de Becker, se recomienda realizar un panel de inmunohistoquímica para distrofinas (Distrofina 1, Distrofina 2 y Distrofina 3) y otras proteínas de superficie del músculo esquelético. Está también el limitante del poco tejido que se puede obtener en dichas biopsias miocárdicas. En el caso de los corazones explantados y autopsias, se encuentran por lo general, un aparato valvular normal, arterias coronarias normales, pero con engrosamiento del miocardio principalmente del ventrículo izquierdo1,5.
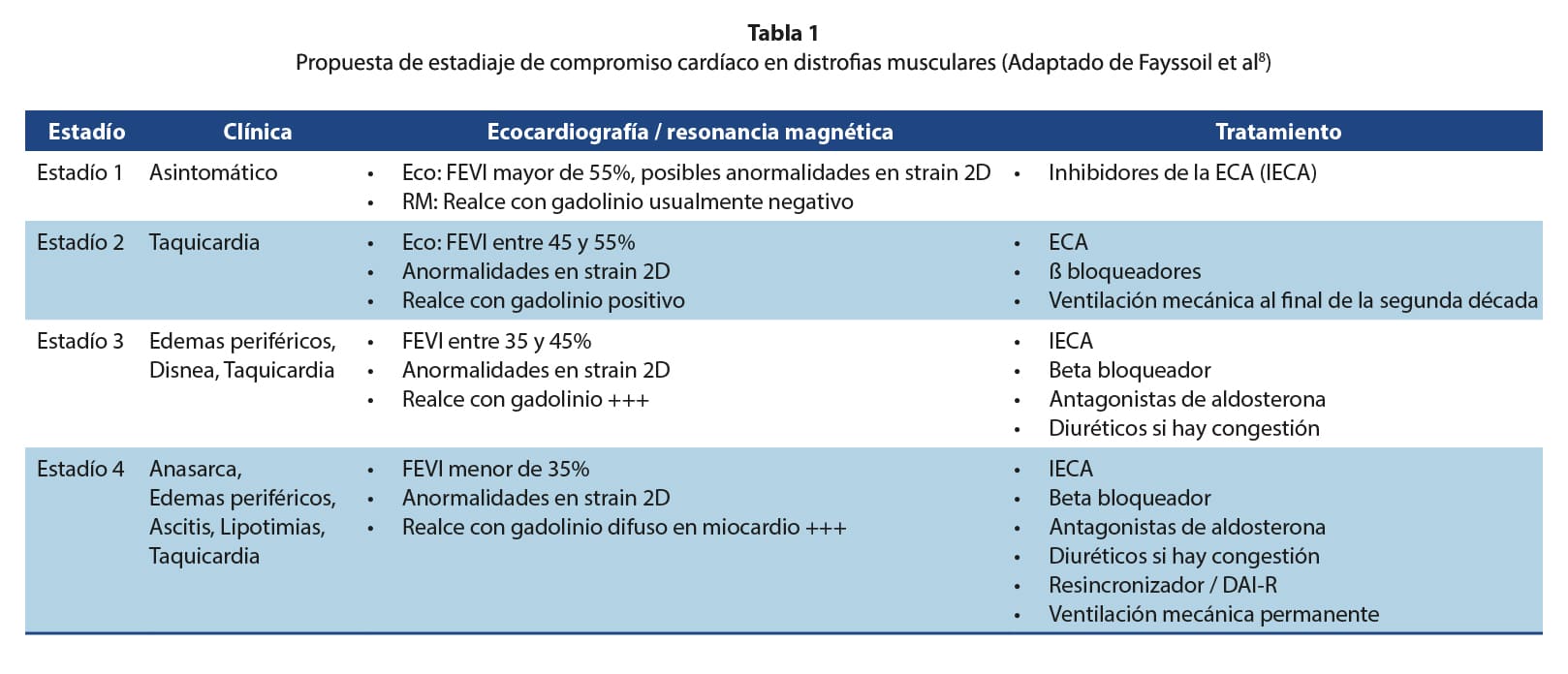
Tomando en cuenta las manifestaciones clínicas, los hallazgos ecocardiográficos y de otros métodos de imágenes, el grupo de trabajo de Fayssoil y cols propuso la estadificación del compromiso cardíaco de los pacientes con distrofia muscular de Duchenne, lo cual guía a su vez, el tratamiento a seguir7. Dado que no existe un sistema de estadiaje específicamente para estos pacientes, esta aproximación puede permitir un abordaje sistemático y un lenguaje universal a la hora de describir el compromiso cardíaco de estos pacientes (Tabla 1)7.
Tratamiento de la afección cardíaca en distrofia muscular
Dado que la afección cardíaca y manifestaciones clínicas del paciente con distrofia muscular se deben a una alteración genética, las opciones terapéuticas son limitadas por el momento. El tratamiento del compromiso cardíaco no varía con la terapia utilizada para manejar otras entidades que presentan falla cardíaca. Se deben evitar todos aquellos fármacos que inducen arritmias, prolongación del QT o desencadenan falla cardíaca.
La terapia de resincronización cardíaca se puede utilizar en pacientes con clases funcionales NYHA III y IV a pesar de tratamiento médico óptimo, reducción severa de la fracción de eyección y retraso de la conducción intraventricular con un QRS mayor a 120 ms3,5. Y en el caso de documentarse arritmias ventriculares, puede plantearse la utilización de un cardiodesfibrilador implantable1.
Los inhibidores de la ECA (IECA) se recomiendan en caso de disfunción diastólica, asimismo, se agregan en caso de disfunción sistólica ventricular izquierda junto con diuréticos y beta bloqueadores. Los casos de falla cardíaca crónica refractaria o intratable pueden ameritar transplante cardíaco, en quienes presentan una clase funcional NYHA III o IV. En el caso particular de los IECA, un estudio realizado en Francia documentó que el uso de estos fármacos en pacientes con distrofia de Duchenne previo a la instauración del daño miocárdico, previene el descenso en la fracción de eyección del ventrículo izquierdo por debajo de 45% a los 60 meses de tratamiento, en pacientes de entre 9 y 13 años de edad8.
El tratamiento con esteroides debe considerarse en estos pacientes con empeoramiento de la función cardíaca, pues se ha mostrado que enlentece la progresión de la disfunción cardíaca, además de prolongar el tiempo de deambulación y mejorar la función de los músculos de la respiración. Incluso se han comparado valores funcionales con resonancia magnética cardíaca, documentando una preservación del tiempo de relajación miocárdica, así como mejoría de la función sistólica ventricular izquierda en pacientes con deflazacort en comparación con placebo9.
Existen reportes desde 1988 de transplante cardíacos ortotópicos en estos pacientes, en los que la evolución ha sido favorable. A pesar de esto, las miopatías hereditarias siguen siendo una contraindicación relativa para trasplante cardíaco, principalmente porque la inmunosupresión necesaria posterior al transplante puede causar progresión de la miopatía y además la disfunción de los músculos respiratorios que puede dificultar la extubación en el postoperatorio3.
El compromiso cardíaco en las distrofias musculares puede presentarse en pacientes con otras manifestaciones extracardíacas o bien en pacientes portadores de la enfermedad, inclusive en mujeres. En estos casos, puede presentarse arritmias, miocardiopatía hipertrófica y dilatada, insuficiencia cardíaca, disfunción diastólica y muerte súbita. Una vez establecido el compromiso cardíaco, debe ser manejado intensivamente al momento del diagnóstico, principalmente para el manejo adecuado de las arritmias y de la falla cardíaca descompensada. Es importante tomar en cuenta que el electrocardiograma es una herramienta importante en estos pacientes, donde hallazgos tempranos del mismo pueden traducir compromiso del ventrículo derecho, por ejemplo; y en algunos casos, como en el aquí presentado, los cambios son más sutiles. El pronóstico en general de estos pacientes es favorable si ésta entidad se reconoce tempranamente y se trata adecuadamente, de lo contrario, más bien influyen en su mortalidad. La utilización de técnicas diagnósticas de imagen principalmente de las no invasivas, pueden permitir un diagnóstico cada vez más temprano de anormalidades cardíacas iniciales, lo cual es de suma importancia para el inicio del tratamiento oportuno.
1. Yilmaz A, Sechtem U. Cardiac involvement in muscular dystrophy: advances in diagnosis and therapy. Heart 2012; 98: 420-429 [PMID: 22311853 DOI: 10.1136/heartjnl-2011-300254]
2. Indorkar, R., Al-Yafi, M., Romano, S., Levin, B. R., & Farzaneh-Far, A. (2017). Cardiomyopathy in muscular dystrophy. QJM: An International Journal of Medicine, 111(4), 267–268. 7
3. Ho R, Nguyen ML, Mather P. Cardiomyopathy in Becker muscular dystrophy: Overview. World J Cardiol 2016; 8(6): 356-361 3
4. Groh, W., Tomaselli, G. and Zipes, D. (2019). Neurologic Disorders and Cardiovascular Disease. In: E. Braunwald, Zipes, Libby and Tomaselli, ed., Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 11th ed. Elsevier, pp.1890-1909.
5. J Finsterer, C Stöllberger. Cardiac involvement in Becker muscular dystrophy. Can J Cardiol 2008;24(10):786-792. 1
6. Saito M, Kawai H, Akaike M, Adachi K, Nishida Y, Saito S. Cardiac dysfunction with Becker muscular dystrophy. Am Heart J 1996; 132: 642-7. 2
7. Fayssoil, A., Abasse, S., & Silverston, K. (2017). Cardiac Involvement Classification and Therapeutic Management in Patients with Duchenne Muscular Dystrophy. Journal of Neuromuscular Diseases, 4(1), 17–23 8
8. Duboc D, Meune C, Lerebours G, et al. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol 2005; 45: 855e7.
9. Mavrogeni, S., Papavasiliou, A., Douskou, M., Kolovou, G., Papadopoulou, E., & Cokkinos, D. V. (2009). Effect of deflazacort on cardiac and sternocleidomastoid muscles in Duchenne muscular dystrophy: A magnetic resonance imaging study. European Journal of Paediatric Neurology, 13(1), 34–40.





{kind=link}